Rosettafold3 for Protein Structure Prediction 🔮
Protein structure prediction has been a fundamental problem in computational biology for decades. Alphafold2's breakthrough in accurately predicting protein structures from sequence alone has revolutionized the field. RoseTTafold provided an open-source alternative with competitive performance.
Alphafold3 and RoseTTafold3 build on their predecessors and extend to biomolecular complexes including DNA, RNA and small molecules.
Structure prediction models reveal unprecedented insights into the function of proteins whose structure has not been resolved experimentally. It is also used in protein design pipelines for in silico screening.
This Space pairs well with the companion RFDiffusion3, LigandMPNN and ESM-2 Spaces for a full design pipeline!
This space allows you to fold bimolecular complexes using Hugging Face's hardware and download the results! It is based on RosettaCommons' foundry repository.
Image and Model Source:Corley N, Mathis S, et al. Accelerating Biomolecular Modeling with AtomWorks and RF3. bioRxiv [Preprint]. 2025 Aug 15:2025.08.14.670328. doi: 10.1101/2025.08.14.670328. PMID: 40832246; PMCID: PMC12363939.
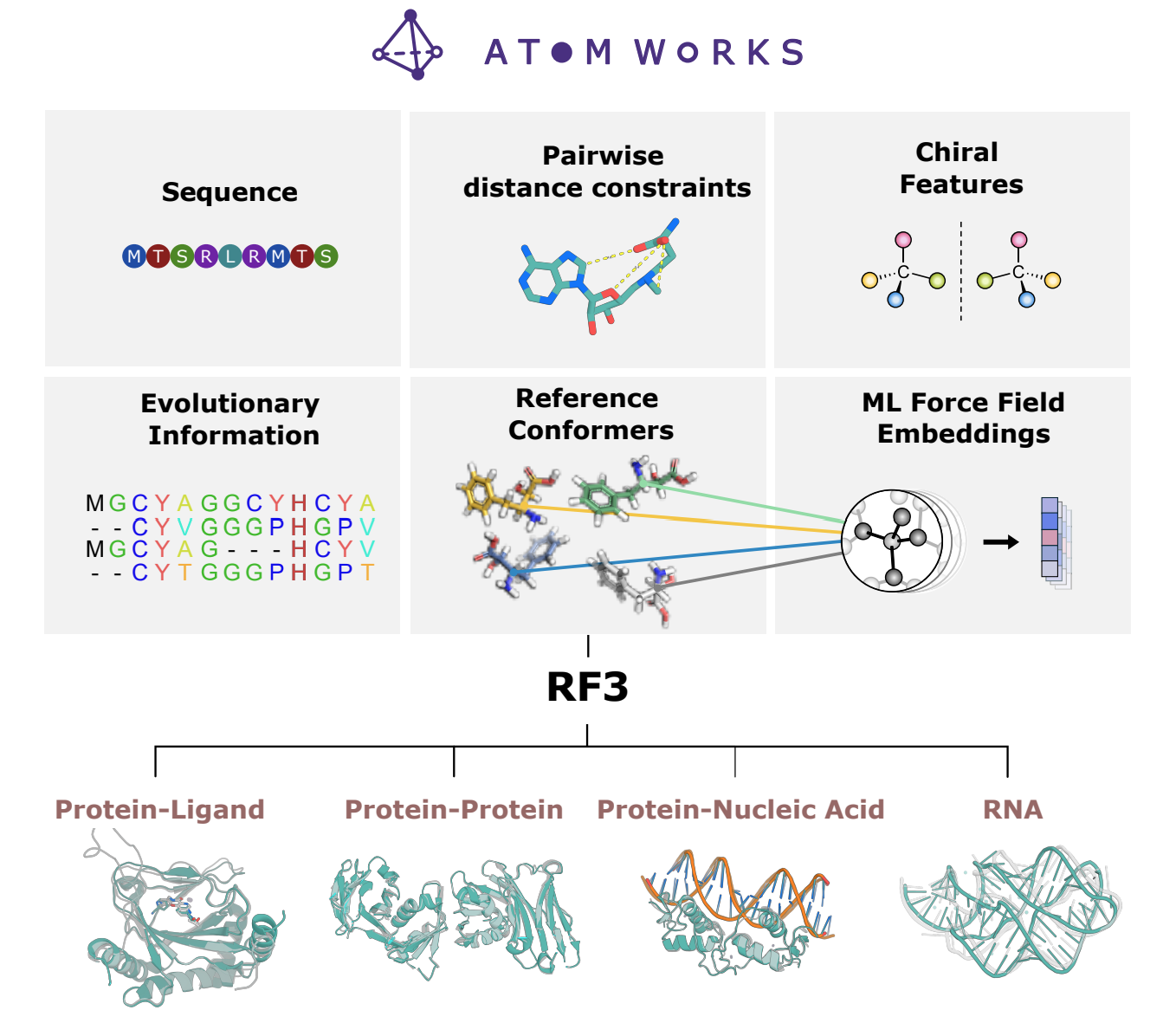{kind=link}
How to Use this Space
Hugging Face spaces is a great tool to build quick machine learning demos and expose interfaces for popular models. This space runs the RF3 model with a convenient interface for input configurations and inference parameters.
The green Running on ZERO at the top of the page indicates that this space runs on Hugging Face's ZeroGPU technology. This means that all jobs are run on NVIDIA H200 GPUs and that the number of GPUs allocated to this space scale automatically based on demand.
Individual jobs On ZeroGPU spaces are limited to 240 seconds for PRO users or members of an organization. Hence, make sure that you are logged in before launching a job to avoid errors and keep the size of the job manageable. The time taken significantly depends on the configuration run. An easy way to reduce it is to reduce the number of diffusion steps, which is set to 200 by default but can be reduced to 100 or 50 without a significant drop in performance. One can also reduce the number of predictions. These are the only command-line parameters available on this interface, along with the early stopping pLDDT threshold. This threshold stops the model if it becomes obvious during the process that the design is not folding properly. It helps save time and resources for erroneous inputs. Its default value of 0.5 is adequate for most use cases. For more information on RosettaFold3's command-line parameters and capabilities, please refer to the documentation.
In this Space, we make the difference between job files and support files.
- Job files are directly fed to the model as inputs. They are typically .json files with detailed descriptions of the folding task, but can also be .pdb or .cif files. In these last two cases,
the model folds all sequences in the file without additional conditioning. It is especially useful for users using this Space for verification of LigandMPNN's designed sequences,
as its
backbonesoutput directory can be directly fed into the space. - Support files are needed when conditioning a job (e.g templating) or using a multiple sequence alignment (MSA), they are mentioned in the .json job files. They can be .pdb or .cif files containing a ground truth structure or a .a3m.gz file containing the MSA.
Example files can be found in the inputs directory of this repo. They are
all taken from RosettaCommons' foundry repository.